46 |
Klinisk Biokemi i Norden · 3 2013
Doktorgradsavhandling:
Tyrosinemi type I
Yngve Thomas Bliksrud
Avdeling for Medisinsk Biokjemi, Oslo Universitetssykehus, Oslo
Yngve Thomas Bliksrud forsvarte
30. november 2012 sin avhandling
”Hereditary tyrosinaemia type I,
Studies on the molecular genetics
and DNA repair enzymes” for gra-
den ph.d. ved Universitetet i Oslo.
Magnar Bjørås, Helge Rootwelt og
Berit Woldseth var veiledere og
arbeidet ble utført ved Avdeling for medisinsk biok-
jemi, OUS, Rikshospitalet.
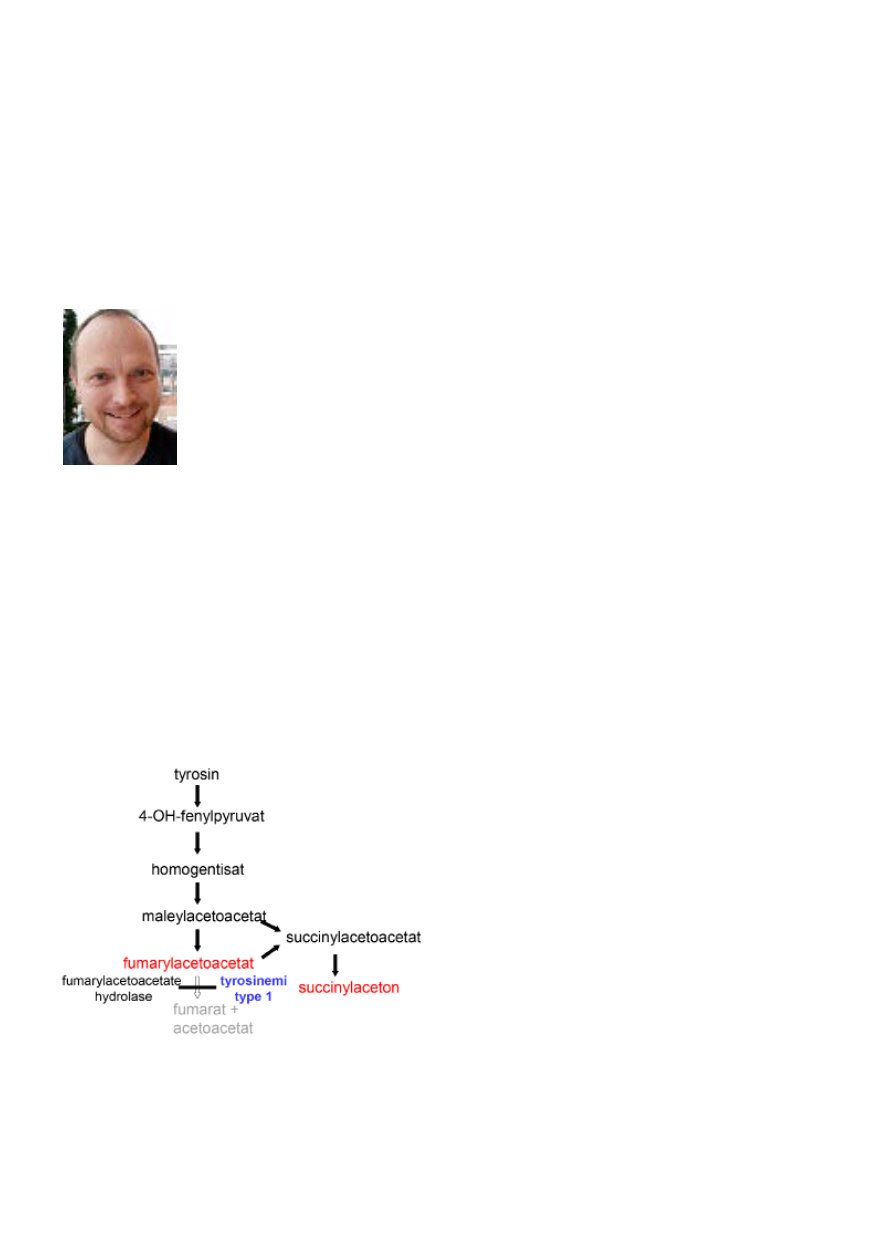
Tyrosinemi type I er en medfødt stoffskiftesykdom
som forekommer litt hyppigere i Norge enn i de fleste
andre land. Den arves autosomalt recessivt, skyldes svikt
i enzymet fumarylacetoacetat hydrolase (FAH) som
katalyserer siste trinn i tyrosinnedbrytningen (se figur).
Blokaden fører til opphopning av skadelige metabolitter
som fumarylacetoacetat (FAA) og succinylaceton (SA).
Sykdommen oppdages i barnealder og rammer i hove-
dsak lever, men også nyrer og nervesystem. Tyrosinemi
type I er assosiert med ustabilt DNA og økt mutasjons-
frekvens i hepatocytter. Den sterkt økte risikoen for
hepatocellulært carcinom (HCC) ved tyrosinemi type
I er et uttrykk for dette. Et annet uttrykk for høy muta-
sjonsfrekvens er et merkverdig fenomen med spontan
korreksjon av medfødte genetiske feil i leverceller hos
tyrosinemipasienter som ble beskrevet i 1994. Levre fra
tyrosinemi type I-pasienter som var tatt ut pga levercel-
lekreft ble undersøkt. I et stort flertall av disse ble det
foruten kreftsvulster påvist cellekloner (ikke kreftceller)
som tilsynelatende var blitt friske av sin tyrosinemi type
I. Enzymet FAH ble påvist ved immunhistokjemi i disse
cellene til forskjell fra vevet omkring. Sekvensering av
FAH-genet i de samme cellene viste at én av de to med-
fødte mutasjonene var korrigert til frisk variant. Den
genetiske korreksjonen har blitt forklart som et tilfeldig
utslag av økt mutasjonsfrekvens i levercellene hos disse
pasientene. Mekanismen bak den økte mutasjonsfrek-
vensen og den høye kreftrisikoen er ufullstendig forstått,
men opphopning av alkylerende metabolitter, oksida-
tivt stress og redusert glutathionkonsentrasjon inngår
i patofysiologien.
Ved vårt laboratorium for utredning av medfødt stoff-
skiftesykdom, som er det eneste i sitt slag i Norge, har
30 norske pasienter fått diagnosen tyrosinemi type I de
siste ca. 30 år. I gradsarbeidet blir DNA-forandringer i
19 av disse pasientene beskrevet, det gis en oversikt over
mutasjonene i den norske befolkningen inkludert tre
små delesjoner som ikke tidligere er beskrevet. Arbei-
det viser at det er i alt minst 9 ulike sykdomsgivende
mutasjoner i den norske befolkningen, og at 65% av
de norske pasientene er sammensatt heterozygote (to
ulike sykdomsgivende mutasjoner). Den noe høyere
forekomsten av tyrosinemi type I i Norge (1 pr 74.800
levendefødte) sammenlignet med de fleste andre land
skyldes dermed et relativt høyt antall sykdomsgivende
mutasjoner i befolkningen. Dette til forskjell fra andre
deler av verden med økt forekomst hvor årsaken er
”founder effects” eller inngifte.
En alternativ forklaring på fenomenet med spontan
Figur 1.
Tyrosin brytes ned i fem trinn hovedsakelig i levercel-
ler. Tyrosinemi type I er en svikt i enzymet FAH som kataly-
serer siste trinn. Svikten fører til opphopning av blant annet
fumarylacetoacetat og succinylaceton.


